Hey, 准备好解锁药物设计的最前沿科技--分子动力学模拟!其能够实时观察分子如何与疾病靶点“对话”,预测它们的每一次亲密接触!这究竟是如何实现的?一文解析~
01
分子模拟的简要介绍
在介绍分子动力学模拟方法之前,我们首先来认识一下分子模拟的概念,分子模拟是一种计算化学方法,它通过计算机程序模拟分子或分子体系的行为,以研究其结构、性质和相互作用。这种方法能够在原子或分子水平上提供对化学过程和物理现象的深入理解,是实验和理论化学的重要补充。
分子模拟主要包括分子力学 (Molecular Mechanics, MM)、蒙特卡洛 (Monte Carlo, MC) 模拟、分子动力学 (Molecular Dynamics, MD) 模拟、量子化学计算、第一性原理分子动力学 (Ab Initio Molecular Dynamics, AIMD)、粗粒化模型 (Coarse-Grained Models) 以及自由能计算等方法 (图 1),其中应用较为广泛的是分子动力学方法和蒙特卡洛方法。与蒙特卡洛模拟相比,分子动力学模拟在多个方面展现出其独特的优势,尤其在处理分子体系的动态行为和时间分辨问题方面。

图 1. 结合分子动力学模拟的虚拟筛选工作流程[1]。
02
分子动力学模拟的作用原理
分子动力学模拟的作用原理是利用经典力学的牛顿运动定律,结合统计力学的概念,通过数值方法模拟原子或分子在给定力场作用下的运动和相互作用。
在模拟开始时,设定分子体系的初始构型和速度,这些初始条件通常基于实验观测或理论预测。随后,模拟中的每个原子或分子根据其所受的力场作用,通过数值积分方法 (如 Verlet 或 Beeman 算法,图 2) 来更新其位置和速度,这些力场包括成键相互作用和非键相互作用,可以通过参数化的势函数来描述。在每个时间步长中,系统的状态 (包括原子的位置和速度) 都会被更新,从而模拟出原子在时间上的动态行为。时间步长的选择对于保证模拟的准确性和稳定性至关重要,它必须足够小以捕捉到体系中的关键物理现象。在模拟过程中,可以对系统的温度和压强进行控制,以模拟不同的热力学条件。通过收集和分析模拟过程中的数据,可以得到分子体系的结构、动力学和热力学性质,如均方位移、相关函数和自由能等。

图 2. 分子动力学基本算法[2]。
03
分子动力学在药物研发中的应用
分子动力学 (MD) 模拟技术不仅能够揭示蛋白质与配体之间复杂的能量相互作用,还为我们提供了深入理解生物大分子动力学结构的丰富信息。MD 模拟已经成为现代药物发现过程中不可或缺的一部分,其应用广泛且成效显著。以下是 MD 模拟在药物发现中的一些具体应用实例。
分子动力学模拟揭示抗病毒化合物与 PrPC “热点”区域的结合、稳定机制
PrPC 的结构转变到 PrPSc (病理型朊蛋白) 是导致传染性海绵状脑病 (即朊疾病) 的主要原因,因此开发能抑制这一转变的有效抗朊病毒药物是一个有希望的治疗方向。
Shuangyan Zhou 等人使用分子动力学模拟方法,研究了几种已知能特异性结合 PrPC“热点”区域并有效稳定其天然结构的抗朊病毒化合物的稳定化机制。结果发现抗朊病毒化合物通过三种机制稳定 PrPC:(1) 稳定 α2 的柔性 C 末端和疏水核心,如 BMD42-29 和 GN8;(2) 仅稳定疏水核心,如 J1 和 GJP49;(3) 通过高结合亲和力稳定 PrPC 的整体结构,如 NPR-056。这些相互作用关键残基的确定对于未来 PrPSc 的药物设计和发现非常重要。

图 3. PrPC 与抗原化合物 BMD42-29、GN8、J1、NPR-056 和 GJP49 之间的详细相互作用[3]。
分子动力学模拟助力 GPCR 药物设计
设计 G 蛋白偶联受体 (GPCR) 变构调节剂是现代药物研究中的一个活跃领域,但由于缺乏这些药物的结合模式和分子机制的知识,这一直是一个挑战。
Ron O. Dror等人利用分子动力学模拟揭示了 M2 毒蕈碱乙酰胆碱受体 (M2 受体) 的几种变构调节剂的结合位点、结合构象和特异性药物-受体相互作用。图 4 还显示了经典配体结合的正、负变构调节机制,包括两个结合位点的耦合构象变化和这些位点上配体之间的静电相互作用。这项研究不仅增进了对 GPCR 变构调节机制的理解,也为开发新型药物提供了重要信息。
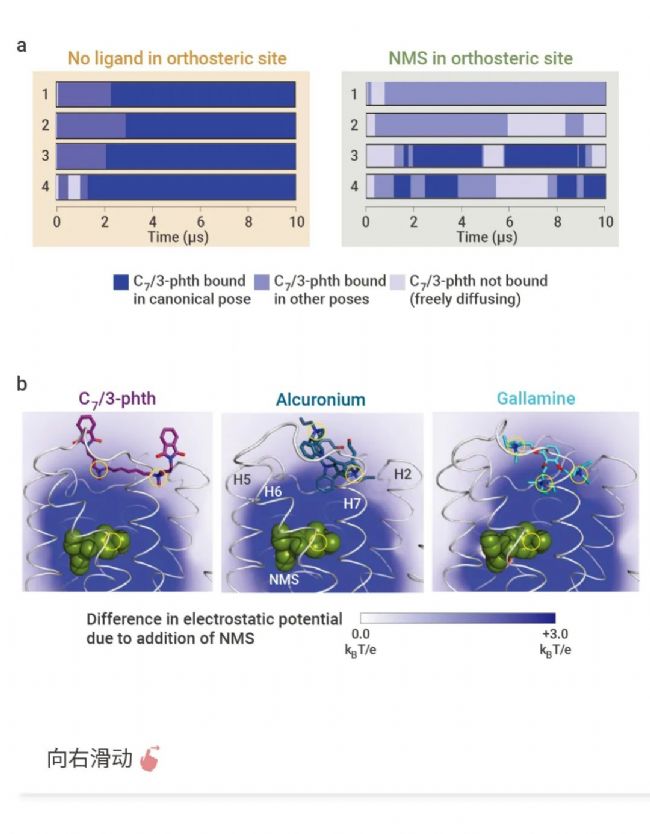

图 4. 正、负变构调节机制[4]。
分子动力学模拟阐明 INSTIs 交叉耐药分子机制
HIV-1 整合酶是治疗 HIV-1 感染的重要药物靶点。然而,对 INSTIs (HIV 整合酶链转移抑制剂) 的交叉耐药性问题日益严重,特别是 E138K/Q148K 双突变体对几种重要的 INSTIs 表现出高水平的耐药性。
Weiwei Xue 等人基于同源建模构建的 HIV-1 整合酶四聚体结构,使用分子动力学 (MD) 模拟和 RIN 分析来研究交叉耐药突变对 INSTIs 的影响。MD 模拟显示,INSTIs 与 HIV-1 整合酶活性位点的结合模式受到 E138K/Q148K 突变的影响,导致活性位点的结构重排 (图 5A、B)。通过结合自由能计算,发现 Pro145 残基在 INSTIs 与 HIV-1 整合酶结合中起到重要作用,特别是其与 INSTIs 的疏水相互作用 (图 5C、D)。该研究不仅为理解 HIV-1 整合酶抑制剂的交叉耐药机制提供了重要信息,而且为未来合理设计新的 INSTIs 以克服交叉耐药性提供了理论基础。
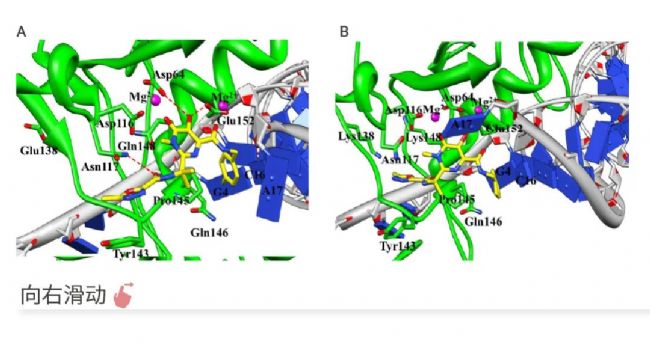

图 5. 分子动力学模拟在阐明 HIV-1 链转移抑制剂交叉耐药分子机制中的应用[5]。
04
小结
今天,小 M 带大家深入了解了分子动力学模拟技术在药物研发领域的应用前景及核心原理。作为一种创新的技术,分子动力学模拟技术将在未来的药物研发中发挥更加关键的作用~
| 产品推荐 |
| Virtual Screening 虚拟筛选 (Virtual Screening,VS) 是基于小分子数据库开展的活性化合物筛选。利用小分子化合物与药物靶标间的分子对接运算,虚拟筛选可快速从几十至上百万分子中,遴选出具有成药性的活性化合物,大大降低实验筛选化合物数量,缩短研究周期,降低药物研发的成本。 |
| MCE 50K Diversity Library (HY-L901) MCE 50K Diversity Library 由 50,000 种类药化合物组成。依据谷本相似性 (Tanimoto Coefficient) 及聚类算法 (Bemis-Murcko) 对上百万化合物进行筛选以确保结构多样性。本多样性库具备新颖性、类药性,化合物结构类型多样、化学空间丰富,库中化合物可重复供应,是新药研发的有力工具,可以广泛地应用于高通量筛选 (HTS) 和高内涵筛选 (HCS)。 |
| 分子动力学模拟 分子动力学 (Molecular Dynamics, MD) 模拟是一种基于牛顿力学,综合了物理、数学和化学等多种学科的计算机模拟方法,用于研究分子体系的运动和相互作用、预测分子体系的行为和结构性质。 |
参考详情:
[1] Liu X, Shi D, Zhou S, Liu H, Liu H, Yao X. Molecular dynamics simulations and novel drug discovery. Expert Opin Drug Discov. 2018 Jan;13(1):23-37.
[2] Goñi JR, Orozco M, Gelpí JL. Molecular dynamics simulations: advances and applications. Adv Appl Bioinform Chem. 2015 Nov 19;8:37-47.
[3] Zhou S, Liu X, An X, Yao X, Liu H. Molecular Dynamics Simulation Study on the Binding and Stabilization Mechanism of Antiprion Compounds to the "Hot Spot" Region of PrPC. ACS Chem Neurosci. 2017 Nov 15;8(11):2446-2456.
[4] Dror RO, Green HF, Valant C, Borhani DW, Valcourt JR, Pan AC, Arlow DH, Canals M, Lane JR, Rahmani R, Baell JB, Sexton PM, Christopoulos A, Shaw DE. Structural basis for modulation of a G-protein-coupled receptor by allosteric drugs. Nature. 2013 Nov 14;503(7475):295-9.
[5] Xue W, Jin X, Ning L, Wang M, Liu H, Yao X. Exploring the molecular mechanism of cross-resistance to HIV-1 integrase strand transfer inhibitors by molecular dynamics simulation and residue interaction network analysis. J Chem Inf Model. 2013 Jan 28;53(1):210-22.