做为一个在科研服务圈摸爬滚打七年之久的老干部,见过形形色色的实验问题,但要说最悲催的,还属一个即将博士毕业的老师,在论文即将投稿之计,偶然发现NCBI数据库将其当做编码基因研究的分子,更新为了非编码基因,这如同一个晴天霹雳砸懵了他,最终换了课题延迟毕业。
如果你觉得上述案例只是个例,那就错了。
以NCBI为例,人基因下辖转录本数量动辄数十个,甚至上百个,但小鼠就没这么多了,其他物种一个基因往往只收录了一个转录本,是人类太高级了?一个基因需要表达很多模板行使功能?答案是否定的,自然面前人与动物是平等的,在基因数量方面,人的少于很多生物,甚至少于昆虫,转录本数量也是一样。
以当前生物圈研究极为深入的分子TP53为例,谷歌以“TP53”为词条搜索,结果达一千五百万条,按理说,其序列应该是确定的了,但在两大核酸数据库里,两者的信息:
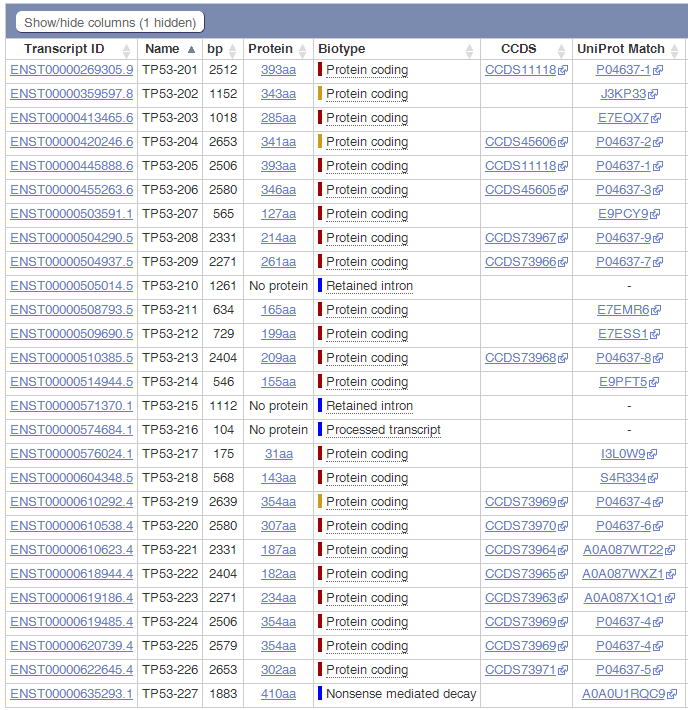
Ensembl数据库收录的TP53转录本数量:
NCBI数据库收录的TP53转录本数量:
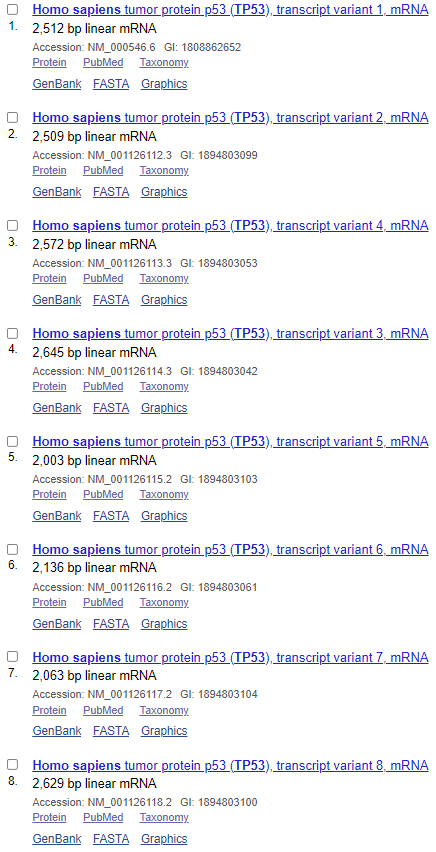
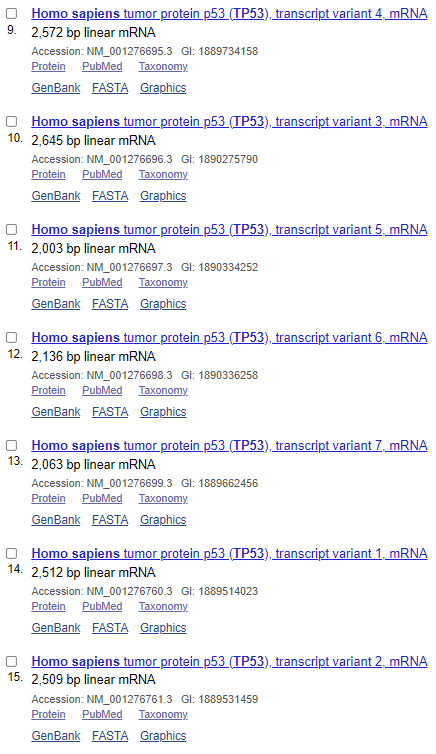
可以看出,在Ensembl数据库中,共收录了27个转录本,三个非编码,二十四个编码;而NCBI收录了十五个编码的转录本,转录本数量就差了接近一倍;再看转录本长度,完全一致的不超过三个;蛋白差异只有一半的转录本是一致的。
想想看,研究透彻程度超过TP53分子的不会超过十个,但是人光编码基因就有2w+,如果连人的基因研究都不够透彻,那其他物种的就没法看了。
当前数据库信息的更新情况
以C9orf62为例,其在2020年八月之前转录本为NM_173520.3,是典型的编码转录本,而现在是NR_171012.1,名称也修正为非编码的名称LINC02907
小鼠Gm10033在2019年四月之前转录本是非编码的NR_038044.1;之后变为编码基因且含有两种不同形式的蛋白转录本NM_001374600.1,NM_001374599.1
在短短的三年时间之内,人、小鼠、大鼠三大物种的基因及转录本条目数更新信息如下:
新增基因17,461个,新增的基因下的新增转录本30,202条
修改基因163,800个,修改的基因下新增了转录本135,739条,修改的基因下修改了转录本268,789条,修改的基因下删除了转录本51,9007条
删除基因98,014个,删除的基因下删除了转录本70,182条
这是个多么恐怖的更新数量,由此,我们对当前数据库的很多基因序列是抱着怀疑的态度审视的。
为何基因会有如此多的转录本呢?
在蛋白数据库UNIPROT的expression处对TP53标注如下:
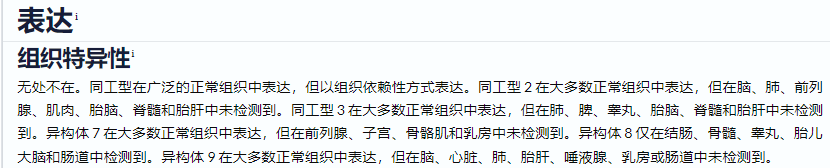
由此可见,虽然一个基因有很多转录本,但这些转录本有明显的组织倾向性或者在限定的发育时间表达,众多的转录本不是一窝蜂表达出来的;此外,一些转录本在特定情况,如免疫,缺氧,精神紧张这些外部刺激的因素下限定表达。
基因or转录本为何出现不准确的情况
测序技术:测序技术在不断的发展,基因组或者基因序列如果存在复杂情况(高/低GC、重复序列等),序列是测不准,测不通的;测出来的序列可能也存在误差;
样本问题:当前数据库收录的数据也是从样本测序得来的,要代表全人类的基因,则需要足够的样本量;不同时期,若数据库测序所用样本不一样,则基因数据肯定也存在差异;
数据库收录基因是否编码属性时,很多以软件预测是否有足够长的编码区而编订,并不是依据RNA测序+蛋白质谱相匹配标定转录本,因此会存在很多编码属性错误的转录本。
基因or转录本信息异常可能导致的后果
基因及转录本是否存在:如果因为测序样本或技术的问题,将原本不存在的分子纳入研究范畴,无疑是水中捞月,白忙活;
功能标注错误:最代表的就是非编码基因标注为编码属性、非编码属性标注为编码基因,这种错误是极为致命的;
启动子错误:启动子是基因表达的重要开关,也是研究的热点,由于转录本在不断更新,有相当多的转录本其5UTR序列在更新前后是存在差异的;而启动子是根据5UTR的第一个碱基作为转录起始位点,通常取其上游2000bp做为研究对象,因此,转录本更新前后的启动子序列也是不一样的,可能对结果造成实际的影响;
3UTR错误:同,转录本序列的频繁更新是一定会波及3UTR,而3UTR一直以来都是研究miRNA、蛋白结合的热点区域,再加上现在RNA修饰如m6A的调控,如果序列不一样,验证结果也是不一样的。
如何避开因数据库信息错误导致的坑
重点来了,作为一位科研工作者,如何避免掉入错误数据的坑呢?
文献支撑:对于有参考文献的,重点核实文献的转录本信息(当然,文献一般不给转录本ID,那就根据引物、编码区长度、蛋白大小等有用的信息确认转录本),寻找数据库跟其一致的转录本;
信息核实:对于没有参考文献的基因,核实不同数据库转录本数量、基因长度、编码属性等信息的异同;
初步验证:针对目的基因进行WB,以确认细胞表达的蛋白大小,从而锁定目标转录本;或更为简便的方法是,针对目的基因的RNA同源区设计引物,进行扩增测序,确认细胞实际表达哪些转录本,再进行挑选研究,避免太多无效转录本的干扰(这也是关键问题,很多老师问,转录本太多,我怎么知道挑选哪个进行研究,所以比较简便的是验证下实际表达的转录本);
对于较新的分子,或者是未曾研究过的分子,无论是编码基因还是非编码基因,对于其是否具有编码能力,可以采取构建质粒验证下,成本不高,周期也很快;
对于启动子、3UTR研究的老师,可以做RACE测序,确认全长的UTR序列;觉得RACE麻烦的话,可以针对目标UTR设计PCR引物,验证是否存在对应的RNA序列(例如:在预测miRNA-3UTR结合时,targetscan经常引用旧数据,取结合位点,但是新的转录本序列不含此结合序列,那么简单的方法就是验证细胞的此基因是否含有此结合序列,就针对结合序列两侧设计PCR引物扩增cdna,能够扩增出,就说明结合序列存在的,数据库信息有误,则可以放心大胆的验证)。
最后
古语有云,尽信书,不如无书,经过小编上面简单的论述,相信大家对基因数据有一定的了解,希望各位在科研的路上,不要太执迷于数据库的信息,凡是抱着怀疑的态度看待问题,一切以实验数据,而不是固定的信息为准,才能少走弯路。
吉凯基因可以提供一站式的基因查询,比对,及各类验证方案的设计、定制服务,有需要的老师,多多咨询!!!